One of the myths surrounding ketogenic diets comes from misunderstanding the role of cortisol — the "stress hormone".
In a previous post, we addressed one of the arguments behind this myth: the idea that to activate gluconeogenesis (to make glucose out of protein), extra cortisol must be recruited. That is just factually incorrect, as we showed in the post.
The other argument, which we address here, is more complex.
Like the previous cortisol myth, it involves a faulty chain of reasoning. Here are the steps:
- Ketogenic diets may raise certain measures of cortisol.
- Chronically elevated cortisol is correlated with metabolic sydrome, and therefore higher cortisol measures may indicate the onset of metabolic syndrome.
- Therefore, ketogenic diets could cause metabolic syndrome.
Metabolic syndrome is a terrible and prevalent problem today. It is that cluster of symptoms most strongly identified with diabetes — excess abdominal fat, high blood sugar, and a particular cholesterol profile — but also correlated with other life-threatening conditions such as heart disease and cancer.
In this post, we're going to explain some of the specifics of cortisol metabolism. We'll show how this argument is vague, and how clarifying it leads to the opposite conclusion. The confusion may all stem from misunderstanding one important fact: different measures of cortisol are not equivalent.
First, though, there is an important reason why the argument doesn't make sense.
We already know that a ketogenic diet effectively treats metabolic syndrome. As we will describe below, it turns out that certain cortisol patterns are strongly linked to metabolic syndrome, and might even be a cause of metabolic syndrome. If the cortisol pattern that develops in response to a ketogenic diet were the kind that was associated with metabolic syndrome, then we would expect people on ketogenic diets to show signs of abdominal fat gain, rising blood sugar, and a worsening cholesterol profile, but we see the opposite. This by itself makes it highly unlikely that ketogenic diets raise cortisol in a harmful way.
In other words, because cortisol regulation is so deeply connected to metabolic syndrome, the fact that ketogenic diets reverse symptoms of metabolic syndrome is itself strong evidence that they improve cortisol metabolism.
In Brief
- There are many different measures of cortisol, because researchers have identified many different processes in cortisol metabolism.
- Increases in some of those measurements are consistently linked to metabolic syndrome, and others are not.
- Some researchers believe that cortisol dysregulation is a key underlying factor in metabolic syndrome.
- The cornerstone of this connection may be the activity of an enzyme, 11β-HSD1. It converts from the inactive form cortisone to the active cortisol.
- In metabolic syndrome, 11β-HSD1 is underactive in liver tissue and overactive in fat tissue. This results in a high rate of cortisol clearance, and low rate of regeneration.
- These symptoms of cortisol dysregulation associated with metabolic syndrome were found to be reversed by a keto diet in a study that made the necessary measurements.
Does a ketogenic diet raise cortisol?

Boston Children's Hospital graphic (with our markup in black). Click for the original.
In a widely-cited study [1], from the Harvard-affiliated Boston Children's Hospital, published in the Journal of the American Medical Association, three different diets were tested: a low-fat diet, a low-carb diet, and a low-glycemic-index diet. The study showed that the different diets had substantially different metabolic effects, with the low-carbohydrate diet having the best results. To our surprise, the researchers then recommended the low-glycemic-index diet instead. As they explained in the accompanying press release:
“The very low-carbohydrate diet produced the greatest improvements in metabolism, but with an important caveat: This diet increased participants' cortisol levels, which can lead to insulin resistance and cardiovascular disease.”
The Boston Children's Hospital then went on to produce a graphic advising patients to follow the low-glycemic-index diet, and giving this as the primary reason not to choose the low-carb diet. Here is that graphic, which we've marked (in black) to show our disagreement. (Click for the full version without our markup.)
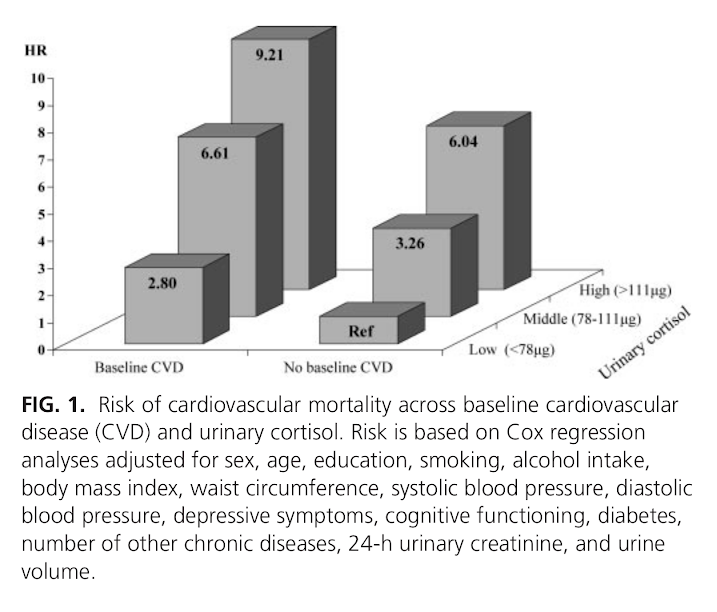
The cortisol levels are an understandable concern, because high urinary cortisol has been epidemiologically associated with a greatly increased risk of death from heart attacks [2].
However, because a ketogenic diet effectively treats metabolic syndrome, we should expect that it also reduces those specific cortisol patterns that are associated with metabolic syndrome (and therefore heart disease). As we show below, this has, in fact, been found.
How is cortisol associated with metabolic syndrome?

Just as we now understand that measuring an individual's total cholesterol without looking at its component parts is inadequate for assessing cardiovascular health, there are different ways to measure cortisol, and only specific patterns of measurements are found with metabolic syndrome.
Cortisol can be measured in fluids, such as urine, saliva, or blood. Within those fluids, the amount of free cortisol can be measured, but so can cortisone, the inactive form, or the metabolites that are the result of enzyme action, and the ratios of any of these to the others can be measured (see Figure 1). Moreover, these measurements have a diurnal rhythm, being higher and lower at different times of the day.
The enzyme 11β-hydroxysteroid dehydrogenase (11β-HSD) can convert back and forth between cortisol and cortisone. 11β-HSD1—a subtype of 11β-HSD—converts cortisone to cortisol. When inactive cortisone is converted to the active cortisol, it is called regeneration. The other enzymes in the illustration break cortisone or cortisol down into metabolites. That process is called clearance. It turns out that measurements of these enzyme are important for evaluating cortisol metabolism.
The cortisol profile that has been associated with metabolic syndrome includes the following characteristics:
- high cortisol production rates [3].
- high cortisol clearance rates [4], [5].
- high 11β-HSD1 expression in fat cells, and low 11β-HSD1 expression in the liver [6], [7], which determines when and where cortisol is regenerated.
Similarly to the way total cholesterol measurement is correlated with heart disease, but only because it is roughly correlated with more informative cholesterol measurements, 24-hour urinary cortisol may be a proxy for production or clearance, but a poor one [3], [4], [7].
Cortisol levels are affected by production, but they are also affected by regeneration and clearance. In other words, if regeneration were increased, or clearance decreased, levels could go up even if production stayed the same or went down. (We previously discussed a similar situation with blood glucose and faulty inference about glucose production rates.) This means that levels can look similar, even when cortisol metabolism is very different.
Implication for those following the “adrenal fatigue” hypothesis: if you measure your cortisol, and it is high, you can't conclude that your adrenal glands are working correspondingly hard. It could be due to increased regeneration and reduced clearance by enzyme activity. Higher cortisol could actually mean the adrenals are working less!In obesity, it appears that production goes up to compensate for high clearance and impaired regeneration, although sometimes not enough to compensate; blood cortisol is sometimes actually lower in obese subjects [8].
How does a ketogenic diet affect the relevant cortisol measures?
In [9], investigators put obese men on either a high-fat/low-carb (fat 66%, carb 4%) or a moderate-fat/moderate-carb (fat 35%, carb 35%) diet ad libitum (eating as much as they wanted). Note that both diets had the same protein percent, and both were lower carb than a standard American diet, but only the high-fat/low-carb diet was at ketogenically low levels.
For the high-fat/low-carb group, “the metabolic syndrome pattern” was reversed: blood cortisol went up, clearance went down, and regeneration went up. This was apparently due to an increase of 11β-HSD1 activity in liver tissue.
(Activity of 11β-HSD1 did not go down in fat tissue of those subjects, but the authors point out that the activity in fat tissue tends to go down when more fat is eaten, and the high-fat/low-carb group weren't actually eating more fat in absolute terms than at baseline, only lower carb.)
This reversal didn't happen in the moderate-fat/moderate-carb group, even though they lost a similar amount of weight.
So the ketogenic diet actually improved the cortisol profile of the participants, making it less like the cortisol profile seen in metabolic syndrome.
Summary
There is some reason to believe that cortisol dysregulation is a key underlying factor in metabolic syndrome [10], [11]. The dysregulation has a particular pattern that seems to be caused by a tissue-specific expression of the enzyme 11β-HSD1.
There is a belief among some researchers that ketogenic diets worsen cortisol metabolism (which could lead to metabolic syndrome and heart disease), but an examination of the specific pattern of cortisol metabolism related to metabolic sydrome shows the opposite.
This is what should have been expected in the first place, since ketogenic diets have already been shown to improve insulin sensitivity (the defining symptom of metabolic syndrome) in repeated randomized controlled trials.
One mechanism by which keto diet improves metabolic syndrome may be its beneficial effect on cortisol metabolism.
Further Reading
For a review of 11β-HSD1, see:
Gathercole LL, Lavery GG, Morgan SA, Cooper MS, Sinclair AJ, Tomlinson JW, Stewart PM.Endocr Rev. 2013 Aug;34(4):525-55. doi: 10.1210/er.2012-1050. Epub 2013 Apr 23.
References:
| [1] | Evidence type: controlled experiment Ebbeling CB, Swain JF, Feldman HA, Wong WW, Hachey DL, Garcia-Lago E, Ludwig DS. JAMA. 2012 Jun 27;307(24):2627-34. doi: 10.1001/jama.2012.6607. (Emphases ours) "Participants Overweight and obese young adults (n=21). "Interventions After achieving 10 to 15% weight loss on a run-in diet, participants consumed low-fat (LF; 60% of energy from carbohydrate, 20% fat, 20% protein; high glycemic load), low-glycemic index (LGI; 40%-40%-20%; moderate glycemic load), and very-low-carbohydrate (VLC; 10%-60%-30%; low glycemic load) diets in random order, each for 4 weeks. … "Hormones and Components of the Metabolic Syndrome (Table 3) "Serum leptin was highest with the LF diet (14.9 [12.1 to 18.4] ng/mL), intermediate with the LGI diet (12.7 [10.3 to 15.6] ng/mL) and lowest with the VLC diet (11.2 [9.1 to 13.8] ng/mL; P=0.0006). Cortisol excretion measured with a 24-hour urine collection (LF: 50 [41 to 60] μg/d; LGI: 60 [49 to 73] μg/d; VLC: 71 [58 to 86] μg/d; P=0.005) and serum TSH (LF: 1.27 [1.01 to 1.60] μIU/mL; LGI: 1.22 [0.97 to 1.54] μIU/mL; VLC: 1.11 [0.88 to 1.40] μIU/mL; P=0.04) also differed in a linear fashion by glycemic load. Serum T3 was lower with the VLC diet compared to the other two diets (LF: 121 [108 to 135] ng/dL; LGI: 123 [110 to 137] ng/dL; VLC: 108 [96 to 120] ng/dL; P=0.006). "Regarding components of the metabolic syndrome, indexes of peripheral (P=0.02) and hepatic (P=0.03) insulin sensitivity were lowest with the LF diet. Serum HDL-cholesterol (LF: 40 [35 to 45] mg/dL; LGI: 45 [41 to 50] mg/dL; VLC: 48 [44 to 53] mg/dL; P<0.0001), triglycerides (LF: 107 [87 to 131] mg/dL; LGI: 87 [71 to 106] mg/dL; VLC: 66 [54 to 81] mg/dL; P<0.0001), and PAI-1 (LF: 1.39 [0.94 to 2.05] ng/mL; LGI: 1.15 [0.78 to 1.71] ng/mL; VLC: 1.01 [0.68 to 1.49] ng/mL; P for trend=0.04) were most favorable with the VLC diet and least favorable with the LF diet. … "Although the very low-carbohydrate diet produced the greatest improvements in most metabolic syndrome components examined here, we identified two potentially deleterious effects of this diet. Twenty-four hour urinary cortisol excretion, a hormonal measure of stress, was highest with the very low-carbohydrate diet. Consistent with this finding, Stimson et al reported increased whole-body regeneration of cortisol by 11β-HSD1 and reduced inactivation of cortisol by 5α-and 5β-reductases over 4 weeks on a VLC vs. a moderate-carbohydrate diet. Higher cortisol levels may promote adiposity, insulin resistance, and cardiovascular disease, as observed in epidemiological studies." Comment: It is ironic that the authors bring up Stimson et al. as an example of a study that corroborates their findings. This is the very study [9] that, in our opinion, exonerates the VLC diet with respect to cortisol. |
| [2] | Evidence type: epidemiological observation Vogelzangs N, Beekman AT, Milaneschi Y, Bandinelli S, Ferrucci L, Penninx BW. J Clin Endocrinol Metab. 2010 Nov;95(11):4959-64. doi: 10.1210/jc.2010-0192. Epub 2010 Aug 25. "Context: The stress hormone cortisol has been linked with unfavorable cardiovascular risk factors, but longitudinal studies examining whether high levels of cortisol predict cardiovascular mortality are largely absent. Objective: The aim of this study was to examine whether urinary cortisol levels predict all-cause and cardiovascular mortality over 6 yr of follow-up in a general population of older persons. Design and Setting: Participants were part of the InCHIANTI study, a prospective cohort study in the older general population with 6 yr of follow-up. Participants: We studied 861 participants aged 65 yr and older. Main Outcome Measure: Twenty-four-hour urinary cortisol levels were assessed at baseline. In the following 6 yr, all-cause and cardiovascular mortality was ascertained from death certificates. Cardiovascular mortality included deaths due to ischemic heart disease and cerebrovascular disease. Results: During a mean follow-up of 5.7 (sd = 1.2) yr, 183 persons died, of whom 41 died from cardiovascular disease. After adjustment for sociodemographics, health indicators, and baseline cardiovascular disease, urinary cortisol did not increase the risk of noncardiovascular mortality, but it did increase cardiovascular mortality risk. Persons in the highest tertile of urinary cortisol had a five times increased risk of dying of cardiovascular disease (hazard ratio = 5.00; 95% confidence interval = 2.02–12.37). This effect was found to be consistent across persons with and without cardiovascular disease at baseline (p interaction = 0.78). Conclusions: High cortisol levels strongly predict cardiovascular death among persons both with and without preexisting cardiovascular disease. The specific link with cardiovascular mortality, and not other causes of mortality, suggests that high cortisol levels might be particularly damaging to the cardiovascular system." |
| [3] | Evidence type: experiment Jonathan Q. Purnell, Steven E. Kahn, Mary H. Samuels, David Brandon, D. Lynn Loriaux, and John D. Brunzell Am J Physiol Endocrinol Metab. 2009 February; 296(2): E351–E357. "Controversy exists as to whether endogenous cortisol production is associated with visceral obesity and insulin resistance in humans. We therefore quantified cortisol production and clearance rates, abdominal fat depots, insulin sensitivity, and adipocyte gene expression in a cohort of 24 men. To test whether the relationships found are a consequence rather than a cause of obesity, eight men from this larger group were studied before and after weight loss. Daily cortisol production rates (CPR), free cortisol levels (FC), and metabolic clearance rates (MCR) were measured by stable isotope methodology and 24-h sampling; intra-abdominal fat (IAF) and subcutaneous fat (SQF) by computed tomography; insulin sensitivity (SI) by frequently sampled intravenous glucose tolerance test; and adipocyte 11β-hydroxysteroid dehydrogenase-1 (11β-HSD-1) gene expression by quantitative RT-PCR from subcutaneous biopsies. Increased CPR and FC correlated with increased IAF, but not SQF, and with decreased SI. Increased 11β-HSD-1 gene expression correlated with both IAF and SQF and with decreased SI. With weight loss, CPR, FC, and MCR did not change compared with baseline; however, with greater loss in body fat than lean mass during weight loss, both CPR and FC increased proportionally to final fat mass and IAF and 11β-HSD-1 decreased compared with baseline. These data support a model in which increased hypothalamic-pituitary-adrenal activity in men promotes selective visceral fat accumulation and insulin resistance and may promote weight regain after diet-induced weight loss, whereas 11β-HSD-1 gene expression in SQF is a consequence rather than cause of adiposity. "Previous studies have shown that compared with women, men have increased CPR (29), cortisol levels (29, 44), and visceral adiposity (9, 13). Given that hypercortisolemia can induce central obesity in disease states such as Cushing's syndrome, elevated endogenous cortisol secretion has been considered a potential mechanism that contributes to the expression of visceral adiposity in humans. However, of four previous reports that used 24-h urinary excretion rates of cortisol as a surrogate for cortisol production, only one found significant relationships between urinary secretion of total glucocorticoids, truncal fat, and insulin sensitivity in men and women (39), while three other studies in men have failed to show associations between urinary glucocorticoid secretion and either WHR (16, 26) or visceral fat (48). These studies, however, did not measure cortisol production directly, did not include blood FC, and did not test for differences in circadian variations of blood levels of cortisol, and in only one study was visceral fat specifically measured. [...] "In summary, we found in men that increased CPR and circulating FC are associated with accumulation of IAF, but not SQF, and with insulin resistance and impaired islet β-cell compensation (DI)." |
| [4] | Evidence type: observational Holt HB, Wild SH, Postle AD, Zhang J, Koster G, Umpleby M, Shojaee-Moradie F, Dewbury K, Wood PJ, Phillips DI, Byrne CD. Diabetologia. 2007 May;50(5):1024-32. Epub 2007 Mar 17. "AIMS/HYPOTHESIS: The regulation of cortisol metabolism in vivo is not well understood. We evaluated the relationship between cortisol metabolism and insulin sensitivity, adjusting for total and regional fat content and for non-alcoholic fatty liver disease. "MATERIALS AND METHODS: "Twenty-nine middle-aged healthy men with a wide range of BMI were recruited. We measured fat content by dual-energy X-ray absorptiometry and magnetic resonance imaging (MRI), liver fat by ultrasound and MRI, the hypothalamic-pituitary-adrenal axis by adrenal response to ACTH(1-24), unconjugated urinary cortisol excretion, corticosteroid-binding globulin, and cortisol clearance by MS. We assessed insulin sensitivity by hyperinsulinaemic-euglycaemic clamp and by OGTT. "RESULTS: "Cortisol clearance was strongly inversely correlated with insulin sensitivity (M value) (r = -0.61, p = 0.002). Cortisol clearance was increased in people with fatty liver compared with those without (mean+/-SD: 243 +/- 10 vs 158 +/- 36 ml/min; p = 0.014). Multiple regression modelling showed that the relationship between cortisol clearance and insulin sensitivity was independent of body fat. The relationship between fatty liver and insulin sensitivity was significantly influenced by body fat and cortisol clearance. "CONCLUSIONS/INTERPRETATION: "Cortisol clearance is strongly associated with insulin sensitivity, independently of the amount of body fat. The relationship between fatty liver and insulin sensitivity is mediated in part by both fatness and cortisol clearance." [...] "Since we showed no strong associations between measures of insulin sensitivity and 09.00 h cortisol levels, ACTH-stimulated cortisol concentrations, and unconjugated urinary cortisol excretion, these findings suggest that the relationship between these other aspects of cortisol metabolism and insulin sensitivity is relatively weak." |
| [5] | Evidence type: experiment Purnell JQ, Brandon DD, Isabelle LM, Loriaux DL, Samuels MH. J Clin Endocrinol Metab. 2004 Jan;89(1):281-7. (emphasis ours) "The present study was designed to examine the hypothesis that hypothalamic-pituitary-adrenal axis activity as measured by 24-h cortisol production rate (CPR) and plasma levels of free cortisol is linked to increased body fat in adults, and that increased cortisol levels with aging results from increased CPR. Fifty-four healthy men and women volunteers with a wide range of body mass indexes and ages underwent measurement of CPR by isotope dilution measured by gas chromatography-mass spectroscopy, cortisol-binding globulin, and free cortisol in pooled 24-h plasma, body composition, and leptin. Cortisol clearance rates were determined from the 10-h disappearance curves of hydrocortisone after steady-state infusion in a separate group of lean and obese subjects with adrenal insufficiency. Although CPR significantly increased with increasing body mass index and percentage body fat, free cortisol levels remained independent of body composition and leptin levels due to increased cortisol clearance rates. CPR and free cortisol levels were, however, significantly higher in men than women. In addition, 24-h plasma free cortisol levels were increased with age in association with increased CPR, independent of body size. This increase in hypothalamic-pituitary-adrenal axis activity may play a role in the alterations in body composition and central fat distribution in men vs. women and with aging." |
| [6] | Evidence type: review of human and non-human animal experiments Espíndola-Antunes D, Kater CE. Arq Bras Endocrinol Metabol. 2007 Nov;51(8):1397-403. "Human studies "The bulk of evidences points both to an overexpression and an increased activity of 11bHSD1 in subcutaneous (SAT) and visceral adipose tissue (VAT) of obese subjects, although biopsies of the omentum were conducted in but a few studies. Several groups have shown higher 11bHSD1 mRNA expression in obese compared to non-obese subjects (29-32), although not all studies agree (33). Direct in vivo measurements using microdialysis in SAT also suggest an increase in the conversion rate of cortisone to cortisol (34). Moreover, 11bHSD1 mRNA expression positively correlates with obesity (body mass index and abdominal circumference), body composition, insulin resistance (30-32), resistins and other cytokines, as TNFa, IL-6, and leptin (35). "The whole body 11bHSD1 activity reflects mainly hepatic expression. Initial studies that relied on measurements of cortisol-to-cortisone metabolites in urine (23,36) should be taken with caution as indicative of 11bHSD1 activity, because several other cortisol and cortisone metabolizing enzymes are deregulated in obesity (36). Of greater importance is the finding of reduced hepatic 11bHSD1 activity measured by the conversion of orally administered cortisone to cortisol (23,37). Thus, 11bHSD1 upregulation in obesity seems not to be a generalized process. In both the whole body and the splanchnic circulation there are no differences between obese and lean subjects regarding cortisol regeneration rates (as measured by [2H4]-cortisol tracer), presumably because an upregulation in adipose tissue is counterbalanced by a downregulation in the liver (15). "Polymorphisms in the 11bHSD1 gene were identified in an attempt to clarify the basis for the increased activity of adipose tissue 11bHSD1 in obesity. In two populations, polymorphisms were associated with an increased risk of diabetes and hypertension, but not obesity (38,39). A polymorphism was also found that predicts lower 11bHSD1 expression and protection against diabetes (40)." |
| [7] | Evidence type: observational Wake DJ, Rask E, Livingstone DE, Söderberg S, Olsson T, Walker BR. J Clin Endocrinol Metab. 2003 Aug;88(8):3983-8. (emphasis ours) "In idiopathic obesity circulating cortisol levels are not elevated, but high intraadipose cortisol concentrations have been implicated. 11beta-Hydroxysteroid dehydrogenase type 1 (11HSD1) catalyzes the conversion of inactive cortisone to active cortisol, thus amplifying glucocorticoid receptor (GR) activation. In cohorts of men and women, we have shown increased ex vivo 11HSD1 activity in sc adipose tissue associated with in vivo obesity and insulin resistance. Using these biopsies, we have now validated this observation by measuring 11HSD1 and GR mRNA and examined the impact on intraadipose cortisol concentrations, putative glucocorticoid regulated adipose target gene expression (angiotensinogen and leptin), and systemic measurements of cortisol metabolism. From aliquots of sc adipose biopsies from 16 men and 16 women we extracted RNA for real-time PCR and steroids for immunoassays. Adipose 11HSD1 mRNA was closely related to 11HSD1 activity [standardized beta coefficient (SBC) = 0.58; P < 0.01], and both were positively correlated with parameters of obesity (e.g. for BMI, SBC = 0.48; P < 0.05 for activity, and SBC = 0.63; P < 0.01 for mRNA) and insulin sensitivity (log fasting plasma insulin; SBC = 0.44; P < 0.05 for activity, and SBC = 0.33; P = 0.09 for mRNA), but neither correlated with urinary cortisol/cortisone metabolite ratios. Adipose GR-alpha and angiotensinogen mRNA levels were not associated with obesity or insulin resistance, but leptin mRNA was positively related to 11HSD1 activity (SBC = 0.59; P < 0.05) and tended to be associated with parameters of obesity (BMI: SBC = 0.40; P = 0.09), fasting insulin (SBC = 0.65; P < 0.05), and 11HSD1 mRNA (SBC = 0.40; P = 0.15). Intraadipose cortisol (142 +/- 30 nmol/kg) was not related to 11HSD1 activity or expression, but was positively correlated with plasma cortisol. These data confirm that idiopathic obesity is associated with transcriptional up-regulation of 11HSD1 in adipose, which is not detected by conventional in vivo measurements of urinary cortisol metabolites and is not accompanied by dysregulation of GR. Although this may drive a compensatory increase in leptin synthesis, whether it has an adverse effect on intraadipose cortisol concentrations and GR-dependent gene regulation remains to be established." |
| [8] | Evidence type: review Brian R. Walker Proceedings of the Nutrition Society / Volume 66 / Issue 01 / February 2007, pp 1-8 (emphasis ours) "The parallels between the clinical features of Cushing’s syndrome and the features of the metabolic syndrome (visceral obesity, hyperglycaemia, hypertension) led to the hypothesis that obesity is associated with glucocorticoid excess (Bjorntorp, 1991). In several monogenic rodent models obesity is accompanied by elevated circulating glucocorticoid levels, and the obesity is prevented by adrenalectomy. Hyperactivity of the HPA axis was thought to reflect chronic stress. However, although there is some evidence for greater stress responsiveness of the HPA axis in obesity (Rosmond et al . 1998; Epel et al . 2000), stress does not appear to explain HPA axis activation in metabolic syndrome (Brunner et al . 2002). Most importantly, in human obesity it appears that cortisol secretion (Marin et al . 1992; Pasquali et al . 1993) is increased to match elevated metabolic clearance (Strain et al . 1982; Andrew et al . 1998; Lottenberg et al . 1998), and does not result in increased plasma cortisol concentrations. Indeed, plasma cortisol concentrations are generally lower amongst obese subjects (Ljung et al . 1996; Walker et al . 2000; Reynolds et al . 2003), i.e. inverse to the effects on the HPA axis seen during starvation (see earlier; p. 2)." |
| [9] | Evidence type: randomised controlled trial Stimson RH, Johnstone AM, Homer NZ, Wake DJ, Morton NM, Andrew R, Lobley GE, Walker BR. J Clin Endocrinol Metab. 2007 Nov;92(11):4480-4. Epub 2007 Sep 4. "CONTEXT: Dietary macronutrient composition influences cardiometabolic health independently of obesity. Both dietary fat and insulin alter glucocorticoid metabolism in rodents and, acutely, in humans. However, whether longer-term differences in dietary macronutrients affect cortisol metabolism in humans and contribute to the tissue-specific dysregulation of cortisol metabolism in obesity is unknown. OBJECTIVE: The objective of the study was to test the effects of dietary macronutrients on cortisol metabolism in obese men. DESIGN: The study consisted of two randomized, crossover studies. SETTING: The study was conducted at a human nutrition unit. PARTICIPANTS: Participants included healthy obese men. INTERVENTIONS, OUTCOME MEASURES, AND RESULTS: Seventeen obese men received 4 wk ad libitum high fat-low carbohydrate (HF-LC) (66% fat, 4% carbohydrate) vs. moderate fat-moderate carbohydrate (MF-MC) diets (35% fat, 35% carbohydrate). Six obese men participated in a similar study with isocaloric feeding. Both HF-LC and MF-MC diets induced weight loss. During 9,11,12,12-[(2)H](4)-cortisol infusion, HF-LC but not MF-MC increased 11beta-hydroxysteroid dehydrogenase type 1 (11beta-HSD1) activity (rates of appearance of cortisol and 9,12,12-[(2)H](3)-cortisol) and reduced urinary excretion of 5alpha- and 5beta-reduced [(2)H](4)-cortisol metabolites and [(2)H](4)-cortisol clearance. HF-LC also reduced 24-h urinary 5alpha- and 5beta-reduced endogenous cortisol metabolites but did not alter plasma cortisol or diurnal salivary cortisol rhythm. In sc abdominal adipose tissue, 11beta-HSD1 mRNA and activity were unaffected by diet. CONCLUSIONS: A low-carbohydrate diet alters cortisol metabolism independently of weight loss. In obese men, this enhances cortisol regeneration by 11beta-HSD1 and reduces cortisol inactivation by A-ring reductases in liver without affecting sc adipose 11beta-HSD1. Alterations in cortisol metabolism may be a consequence of macronutrient dietary content and may mediate effects of diet on metabolic health." |
| [10] | Evidence type: authority (review article) Pereira CD, Azevedo I, Monteiro R, Martins MJ. Diabetes Obes Metab. 2012 Oct;14(10):869-81. doi: 10.1111/j.1463-1326.2012.01582.x. Epub 2012 Mar 8. (emphasis ours) "Recent evidence strongly argues for a pathogenic role of glucocorticoids and 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) in obesity and the metabolic syndrome, a cluster of risk factors for atherosclerotic cardiovascular disease and type 2 diabetes mellitus (T2DM) that includes insulin resistance (IR), dyslipidaemia, hypertension and visceral obesity. This has been partially prompted not only by the striking clinical resemblances between the metabolic syndrome and Cushing’s syndrome (a state characterized by hypercortisolism that associates with metabolic syndrome components) but also from monogenic rodent models for the metabolic syndrome (e.g. the leptin-deficient ob/ob mouse or the leptin-resistant Zucker rat) that display overall increased secretion of glucocorticoids. However, systemic circulating glucocorticoids are not elevated in obese patients and/or patients with metabolic syndrome. The study of the role of 11β-HSD system shed light on this conundrum, showing that local glucocorticoids are finely regulated in a tissue-specific manner at the pre-receptor level. The system comprises two microsomal enzymes that either activate cortisone to cortisol (11β-HSD1) or inactivate cortisol to cortisone (11β-HSD2). Transgenic rodent models, knockout (KO) for HSD11B1 or with HSD11B1 or HSD11B2 overexpression, specifically targeted to the liver or adipose tissue, have been developed and helped unravel the currently undisputable role of the enzymes in metabolic syndrome pathophysiology, in each of its isolated components and in their prevention. In the transgenic HSD11B1 overexpressing models, different features of the metabolic syndrome and obesity are replicated." |
| [11] | Evidence type: non-human animal experiment Schnackenberg CG, Costell MH, Krosky DJ, Cui J, Wu CW, Hong VS, Harpel MR, Willette RN, Yue TL. Biomed Res Int. 2013;2013:427640. doi: 10.1155/2013/427640. Epub 2013 Mar 18. (emphasis ours) "Metabolic syndrome is a constellation of risk factors including hypertension, dyslipidemia, insulin resistance, and obesity that promote the development of cardiovascular disease. Metabolic syndrome has been associated with changes in the secretion or metabolism of glucocorticoids, which have important functions in adipose, liver, kidney, and vasculature. Tissue concentrations of the active glucocorticoid cortisol are controlled by the conversion of cortisone to cortisol by 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1). Because of the various cardiovascular and metabolic activities of glucocorticoids, we tested the hypothesis that 11β-HSD1 is a common mechanism in the hypertension, dyslipidemia, and insulin resistance in metabolic syndrome. In obese and lean SHR/NDmcr-cp (SHR-cp), cardiovascular, metabolic, and renal functions were measured before and during four weeks of administration of vehicle or compound 11 (10 mg/kg/d), a selective inhibitor of 11β-HSD1. Compound 11 significantly decreased 11β-HSD1 activity in adipose tissue and liver of SHR-cp. In obese SHR-cp, compound 11 significantly decreased mean arterial pressure, glucose intolerance, insulin resistance, hypertriglyceridemia, and plasma renin activity with no effect on heart rate, body weight gain, or microalbuminuria. These results suggest that 11β-HSD1 activity in liver and adipose tissue is a common mediator of hypertension, hypertriglyceridemia, glucose intolerance, and insulin resistance in metabolic syndrome." |
No comments:
Post a Comment